
URL: https://www.desy.de/news/news_search/index_eng.html
Breadcrumb Navigation
DESY News: New method opens crystal clear views of biomolecules
News
News from the DESY research centre
New method opens crystal clear views of biomolecules
A scientific breakthrough gives researchers access to the blueprint of thousands of molecules of great relevance to medicine and biology. The novel technique, pioneered by a team led by DESY scientist Professor Henry Chapman from the Center for Free-Electron Laser Science CFEL and reported this week in the scientific journal Nature, opens up an easy way to determine the spatial structures of proteins and other molecules, many of which are practically inaccessible by existing methods. The structures of biomolecules reveal their modes of action and give insights into the workings of the machinery of life. Obtaining the molecular structure of particular proteins, for example, can provide the basis for the development of tailor-made drugs against many diseases. “Our discovery will allow us to directly view large protein complexes in atomic detail,” says Chapman, who is also a professor at the University of Hamburg and a member of the Hamburg Centre for Ultrafast Imaging CUI.

Slightly disordered crystals of complex biomolecules like that of the photosystem II molecule shown here produce a complex continous diffraction pattern (right, the disorder is greatly exaggerated) under X-ray light that contains far more information than the so-called Bragg peaks of a strongly ordered crystal alone (left). Credit: DESY, Eberhard Reimann
In crystallography, the structure of a crystal and of its constituents can be investigated by shining X-rays on it. The X-rays scatter from the crystal in many different directions, producing an intricate and characteristic pattern of numerous bright spots, called Bragg peaks (named after the British crystallography pioneers William Henry and William Lawrence Bragg). The positions and strengths of these spots contain information about the structure of the crystal and of its constituents. Using this approach, researchers have already determined the atomic structures of tens of thousands of proteins and other biomolecules.
But the method suffers from two significant barriers, which make structure determination extremely difficult or sometimes impossible. The first is that the molecules must be formed into very high quality crystals. Most biomolecules do not naturally form crystals. However, without the necessary perfect, regular arrangement of the molecules in the crystal, only a limited number of Bragg peaks are visible. This means the structure cannot be determined, or at best only a fuzzy “low resolution” facsimile of the molecule can be found. This barrier is most severe for large protein complexes such as membrane proteins. These systems participate in a range of biological processes and many are the targets of today's drugs. Great skill and quite some luck are needed to obtain high-quality crystals of them.
“Extreme Sudoku in three dimensions”
The second barrier is that the structure of a complex molecule is still extremely difficult to determine, even when good diffraction is available. “This task is like extreme Sudoku in three dimensions and a million boxes, but with only half the necessary clues,” explains Chapman. In crystallography, this puzzle is referred to as the phase problem. Without knowing the phase – the lag of the crests of one diffracted wave to another – it is not possible to compute an image of the molecule from the measured diffraction pattern. But phases can’t be measured. To solve the tricky phase puzzle, more information must be known than just the measured Bragg peaks. This additional information can sometimes be obtained by X-raying crystals of chemically modified molecules, or by already knowing the structure of a closely-related molecule.
When thinking about why protein crystals do not always “diffract”, Chapman realised that imperfect crystals and the phase problem are linked. The key lies in a weak “continuous” scattering that arises when crystals become disordered. Usually, this non-Bragg, continuous diffraction is thought of as a nuisance, although it can be useful for providing insights into vibrations and dynamics of molecules. But when the disorder consists only of displacements of the individual molecules from their ideal positions in the crystal then the “background” takes on a much more complex character – and its rich structure is anything but diffuse. It then offers a much bigger prize than the analysis of the Bragg peaks: the continuously-modulated “background” fully encodes the diffracted waves from individual “single” molecules.
“If you would shoot X-rays on a single molecule, it would produce a continuous diffraction pattern free of any Bragg spots,” explains lead author Dr. Kartik Ayyer from Chapman's CFEL group at DESY. “The pattern would be extremely weak, however, and very difficult to measure. But the ‘background’ in our crystal analysis is like accumulating many shots from individually-aligned single molecules. We essentially just use the crystal as a way to get a lot of single molecules, aligned in common orientations, into the beam.” With imperfect, disordered crystals, the continuous diffraction fills in the gaps and beyond the Bragg peaks, giving vastly more information than in normal crystallography. With this additional gain in information, the phase problem can be uniquely solved without having to resort to other measurements or assumptions. In the analogy of the Sudoku puzzle, the measurements provide enough clues to always arrive at the right answer.
The best crystals are imperfect crystals
This novel concept leads to a paradigm shift in crystallography — the most ordered crystals are no longer the best to analyse with the novel method. Instead, the best crystals are imperfect crystals. “For the first time we have access to single molecule diffraction – we have never had this in crystallography before,” he explains. “But we have long known how to solve single-molecule diffraction if we could measure it.” The field of coherent diffractive imaging, spurred by the availability of laser-like beams from X-ray free-electron lasers, has developed powerful algorithms to directly solve the phase problem in this case, without having to know anything at all about the molecule. “You don’t even have to know chemistry,” says Chapman, “but you can learn it by looking at the three-dimensional image you get.”
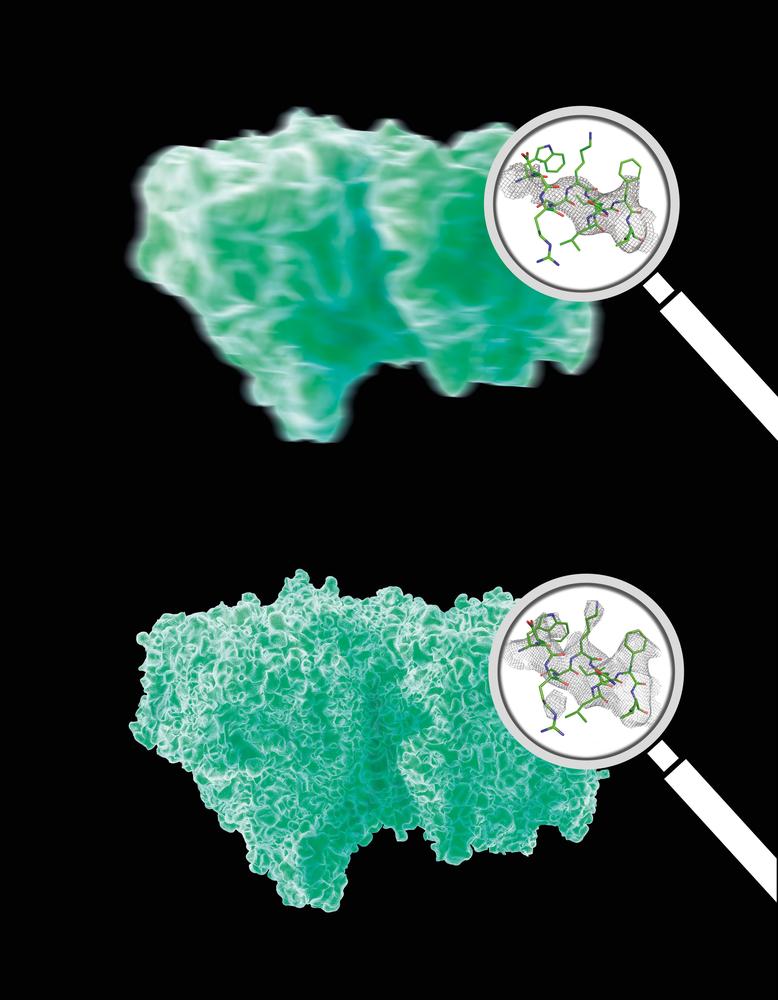
The analysis of the Bragg peaks alone (top) reveals far less details than the analysis of the continuous diffraction pattern (bottom). Magnifying glasses show real data. Credit: DESY, Eberhard Reimann
Including the continuous diffraction pattern into the analysis immediately improved the spatial resolution around a quarter from 4.5 Ångström to 3.5 Ångström (an Ångström is 0.1 nanometres). The obtained image gave fine definition of molecular features that usually require fitting a chemical model to see. “That is a pretty big deal for biomolecules,” explains co-author Dr. Anton Barty from DESY. “And we can further improve the resolution if we take more patterns.” The team had only a few hours of measuring time for these experiments, while full-scale measuring campaigns usually last a couple of days.
The scientists hope to obtain even clearer and higher resolution images of photosystem II and many other macromolecules with their new technique. “This kind of continuous diffraction has actually been seen for a long time from many different poorly-diffracting crystals,” says Chapman. “It wasn’t understood that you can get structural information from it and so analysis techniques suppressed it. We’re going to be busy to see if we can solve structures of molecules from old discarded data.”
Reference:
Macromolecular diffractive imaging using imperfect crystals; Kartik Ayyer et al.; Nature (2016); DOI: 10.1038/nature16949
